Лишняя хромосома у человека. хромосомные аномалии
Содержание:
- Оценка частоты хромосомных нарушений в момент зачатия
- Причины возникновения
- Болезни и расстройства
- Строение хромосомы
- Диагностика синдрома Клайнфельтера
- Что такое анеуплоидия, трисомия, транслокация, мозаицизм
- Ранние признаки
- Причины отклонений, как они проявляются и какова вероятность возникновения
- Как воспитывать ребёнка с хромосомным заболеванием?
- Диагностика
- Причины генетической патологии
- Лечится ли синдром дауна
Оценка частоты хромосомных нарушений в момент зачатия
Можно попробовать расчитать количество зигот с хромосомными аномалиями при зачатии, основываясь на частоте хромосомных аномалий, обнаруживаемых в материале выкидышей. Однако прежде всего следует отметить, что поразительное сходство результатов исследований материала выкидышей, проведенное в разных частях света, говорит о том, что хромосомные сбои в момент зачатия являются очень характерным явлением в репродукции у человека. Кроме того, можно констатировать, что реже всего встречающиеся аномалии (например, трисомии A, B и F) связаны с остановкой развития на очень ранних стадиях.
Анализ относительной частоты различных аномалий, возникающих при нерасхождении хромосом в процессе мейоза, позволяет сделать следующие важные выводы:
1. Единственной моносомией, обнаруживаемой в материале выкидышей, является моносомия X (15% всех аберраций). Напротив, аутосомные моносомии практически не обнаруживаются в материале выкидышей, хотя теоретически их должно быть столько же, сколько и аутосомных трисомий.
2. В группе аутосомных трисомий частота трисомий разных хромосом значительно варьирует. Исследования, выполненные с использованием метода G-бэндинга, позволили установить, что все хромосомы могут быть участницами трисомии, однако некоторые трисомии встречаются гораздо чаще, например, трисомия 16 встречается в 15% случаев всех трисомий.
Из этих наблюдений можно сделать вывод, что, скорее всего, частота нерасхождения разных хромосом приблизительно одинакова, а различная частота аномалий в материале выкидышей связана с тем, что отдельные хромосомные аберрации приводят к остановке развития на очень ранних стадиях и поэтому с трудом поддаются обнаружению.
Эти соображения позволяют приблизительно расчитать реальную частоту хромосомных нарушений в момент зачатия. Расчеты, сделанные Буэ, показали, что каждое второе зачатие дает зиготу с хромосомными аберрациями.
Данные цифры отражают среднюю частоту хромосомных аберраций при зачатии в популяции. Однако данные цифры могут значительно колебаться у разных супружеских пар. У некоторых супружеских пар вероятность возникновения хромосомных аберраций в момент зачатия значительно превышает средний риск в популяции. У таких супружеских пар невынашивание беременности на малых сроках происходит гораздо чаще, чем у остальных супружеских пар.
Данные расчеты подтверждаются другими исследованиями, проведенными с использованием других методов:
1. Классическими исследованиями Хертига
2. Определением уровня хорионического гормона (ХГ) в крови женщин после 10 после зачатия. Часто этот тест оказывается положительным, хотя менструация приходит вовремя или с небольшой задержкой, и субъективно наступления беременности женщина не замечает («биохимическая беременность»)
3. Хромосомный анализ материала, полученного при искусственных абортах показал, что при абортах на сроке 6—9 недель (4—7 недель после зачатия) частота хромосомных аберраций составляет примерно 8%, а при искусственных абортах на сроке 5 недель (3 недели после зачатия) эта частота возрастает до 25%.
4. Было показано, что нерасхождение хромосом в процессе сперматогенеза является очень частым явлением. Так Пирсон и сотр. обнаружили, что вероятность нерасхождения в процессе сперматогенеза для 1-й хромосомы составляет 3,5%, для 9-й хромосомы — 5%, для Y-хромосомы — 2%. Если и другие хромосомы имеют вероятность нерасхождения примерно такого же порядка, то тогда только 40% всех сперматозоидов имеют нормальный хромосомный набор.
Причины возникновения
Во всех случаях оба родителя передают свои гены детям. Трисомия 21 наиболее распространенное генетическое, наследственное заболевание. Эти гены переносятся в хромосомах. Когда клетки ребенка развиваются, каждая клетка должна получать хромосомный набор из 23 пары, всего 46 хромосом. Половина хромосом – от матери, половина – от отца.
Одна из хромосом не отделяется должным образом. Ребенок рождается с тремя копиями или дополнительной частичной копией хромосомы 21 вместо двух. 47 хромосома вызывает проблемы по мере развития мозга и физических функций.
Не существует окончательных научных исследований, свидетельствующих о том, что состояние обусловлено факторами окружающей среды или действиями родителей до или во время беременности.
Открыл, описал его английский врач XIX века Лэнгдон Даун. По иронии судьбы, он не был первым, кто описал это состояние, мало добавил к нашему знанию об этом, и, с большой ошибкой, приписал условие «возвращению» к монголоидной расе.
Расстройство было однажды названо монголизмом, термин, который теперь считается устаревшим.
Генетическая причина болезни обнаружена в 1957 году – трисомия по 21 хромосоме.
Хромосомная болезнь человека (синдром дауна) была изучена с помощью метода кариотипа, когда стало возможным выявлять аномалии хромосомного числа.
В 1971 г. на Парижской конференции была утверждена специальная номенклатура записи кариотипа человека.
Нормальный кариотип человека:
46,ХХ – женщина; 46, ХУ – мужчина.
Кариотип при трисомиях по аутосомам:
47,ХХ,+21 или 47,ХУ,+21 – трисомия по 21 хромосоме (синдром Дауна);
Наследуется ли синдром дауна
В некоторых случаях, он передается по наследству. По данным Национального общества NDSS, около 1 из 700 детей рождаются с ним.
В глобальном масштабе, по состоянию на 2010 год, он происходит примерно 1 на 1000 родов и приводит к примерно 17 000 смертей. Больше таких детей рождаются в странах, где аборт не разрешен, и в странах, где беременность возникает в очень зрелом возрасте.
Число беременностей с аномалией более чем в два раза больше при частых спонтанных выкидышах. Это является причиной 8% всех врожденных расстройств.
Болезни и расстройства
Ниже перечислены некоторые заболевания, связанные с генами 5-й хромосомы, а также гены, дефекты которых вызывают эти заболевания:
- GM2-ганглиозидоз в AB-варианте (англ. GM2-gangliosidosis, AB variant) — GM2A;
- ателостеогенез типа II (англ. atelosteogenesis, type II) — SLC26A2;
- ахондрогенез типа IB (англ. achondrogenesis, type IB) — SLC26A2;
- болезнь Паркинсона;
- болезнь Сандхоффа — HEXB;
- гомоцистинурия (англ. homocystinuria);
- дефицит 3-метилкротонил-КоА-карбоксилазы (англ. 3-methylcrotonyl-CoA carboxylase deficiency) — MCCC2;
- гранулярная дистрофия роговицы типа I и типа II — TGFBI;
- диастрофическая дисплазия (англ. diastrophic dysplasia) — SLC26A2;
- дистрофия боуменовой мембраны роговицы типа I и типа II — TGFBI;
- никотиновая зависимость;
- первичный дефицит карнитина (англ. primary carnitine deficiency) — SLC22A5;
- множественная эпифизарная дисплазия аутосомно-рецессивного типа — SLC22A5;
- семейный аденоматозный полипоз (англ. familial adenomatous polyposis) — APC;
- синдром Коккейна типа A — ERCC8;
- синдром Корнелии де Ланге — NIPBL;
- синдром кошачьего крика — CTNND2, SEMA5A, TERT;
- синдром Сотоса — NSD1;
- синдром Тричера Коллинза — TCOF1;
- синдром Ушера типа 2C — GPR98;
- синдром Элерса — Данлоса с дерматоспараксисом (типа 7C) — ADAMTS2;
- спинальная мышечная атрофия — SMN1 и SMN2.
Хромосомные болезни
Некоторые расстройства вызываются изменениями в структуре или количестве копий 5-й хромосомы:
- синдром кошачьего крика — в большинстве случаев терминальная делеция (с утратой от трети до половины, реже полная утрата) короткого плеча хромосомы, менее чем 10 % случаев причиной являются другие редкие цитогенетические аберрации (например, интерстициальные делеции, мозаицизм, кольца и транслокации); для развития клинической картины синдрома имеет значение не величина утраченного участка, а конкретный незначительный фрагмент хромосомы: потеря небольшой области в полосе 5p15.2 соотносится со всеми клиническими признаками синдрома за исключением характерного плача ребёнка, напоминающего кошачий крик, который отображается на полосу 5p15.3;
- семейный аденоматозный полипоз (англ. familial adenomatous polyposis) — делеция гена опухолевого супрессора (англ. tumor suppressor gene) APC на длинном плече хромосомы (локус 5q21—q22); без полной колектомии (англ. сolectomy) заболевание практически неизбежно приводит к развитию рака толстой кишки;
- задержка роста и развития, развитие характерных черт лица, врожденные дефекты и другие медицинские проблемы — дополнительный участок короткого или длинного плеча хромосомы (частичная 5p или 5q), потеря участка длинного плеча хромосомы (частичная 5q) или образование кольцевой хромосомы (англ. ring chromosome).
Строение хромосомы
Выяснив сколько хромосом у человека, рассмотрим основы их строения. Хромосома является палочковидной структурой, которая состоит из двух сестринских хроматид. Они удерживаются центромерой, располагающейся в области первичной перетяжки. Каждая из хроматид строится из хроматиновых петель. Сам хроматин не подвергается репликации, в отличие от ДНК. С началом этого процесса прекращается синтез РНК. При этом хромосомы находятся в организме в двух состояниях:
- конденсированном (неактивное);
- деконденсированном (активное).
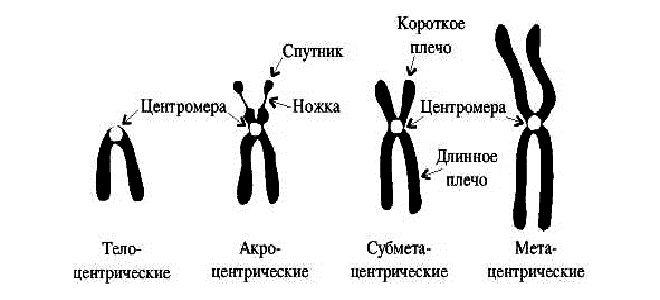
В зависимости от строения генетики выделяют следующие виды хромосом:
- телоцентрические;
- акроцентрические – второе плечо короткое и практически незаметное;
- субметацентрические – внешне напоминают букву L;
- метацентрические – плечики равной длины.
Гомологичные хромосомы
Парные хромосомы человека принято называть гомологичными. При зачатии одна хромосома наследуется от отца, вторая – от матери. На гомологичных хромосомах располагаются гены, которые отличаются по строению, однако выполняют одинаковую функцию. Гомологичные хромосомы имеют схожую последовательность нуклеотидов. Такие хромосомы, расположенные в диплоидных клетках, имеют одинаковые гены. Количество наборов гомологичных хромосом обозначается термином «плоидность». В половых клетках она равна одному (1n), в соматических – двум (2n).
Негомологичные хромосомы
Негомологичные хромосомы – это структуры, которые содержат несхожие гены. Данные структурные элементы не подвергаются конъюгации в процессе мейоза. Негомологичные хромосомы независимо друг от друга комбинируются в клетке. Этот факт был доказан в процессе изучения характеристик наследования признаков путем использования прямого цитологического метода.
Диагностика синдрома Клайнфельтера
Во многих странах синдром Клайнфельтера часто диагностируется ещё до рождения ребёнка, так как многие женщины позднего детородного возраста, в связи с высоким риском генетических дефектов у будущего потомства, используют пренатальную генетическую диагностику плода. Нередко пренатальное выявление синдрома Клайнфельтера является поводом для прерывания беременности, в том числе и по рекомендации врачей. В России анализ кариотипа будущего ребёнка проводится крайне редко.
При подозрении на синдром Клайнфельтера проводят лабораторный анализ крови для определения уровня мужских половых гормонов. Необходима дифференциальная диагностика с другими заболеваниями, протекающими с проявлениями андрогенной недостаточности. Точный диагноз синдрома Клайнфельтера ставят на основании изучения кариотипа (набора хромосом) больного.
Что такое анеуплоидия, трисомия, транслокация, мозаицизм
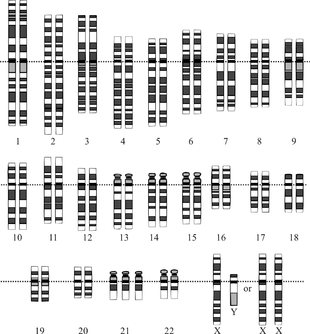
В каждой клетке человеческого организма находится 46 хромосом, в которых выделяют две группы: 22 пары аутосом (пронумерованных с 1 по 22, в зависимости от размера) и пара половых хромосом (XX у женщин, XY у мужчин). Каждая хромосома в паре является гомологичной другой хромосоме в паре.
В норме человек имеет диплоидный набор хромосом, то есть в каждой клетке содержится двойной комплект каждой из 23 хромосом.
Но есть ситуации, в которых клетки содержат ненормальный, не кратный 46, набор хромосом, что называется анеуплоидией. Анеуплоидия может выражаться, например, в наличии добавочной хромосомы (n + 1, 2n + 1 и т. п.) или в нехватке какой-либо хромосомы (n — 1, 2n — 1 и т. п.).
Формы анеуплоидии:
- моносомия (наличие одной из пары хромосом, например, синдром Шерешевского-Тернера, выражающийся в наличие одной половой Х-хромосомы)
- трисомия (наличие трех вместо 2 хромосом пары).
- тетрасомия (4 гомологичные хромосомы вместо пары в диплоидном наборе)
- пентасомия (5 вместо 2-х) встречаются чрезвычайно редко.
Дальше речь пойдет о самых частых хромосомных аномалиях — трисомиях. В некоторых случаях дополнительная хромосома представлена целой отдельной хромосомой (полная трисомия), а в некоторых этот генетический материал переносится на другую хромосому, что называют транслокацией.
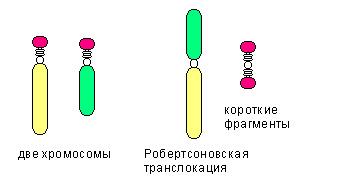
Среди транслокаций также выделяют:
- реципроктную транслокацию, когда неготомологичные хромосомы обмениваются участками
- робертсоновскую транслокацию (см.рис), при которой две неготомологичные хромосомы объединяются в одну.
- Сбалансированная транслокация не сопровождается утратой генетического материала.
Мозаицизмом называют ситуацию, когда среди всех клеток организма есть нормальные, а есть клетки с патологией (например, с трисомией). В этом случае степень отклонений зависит от количества клеток, которые имеет ненормальный генетический материал.
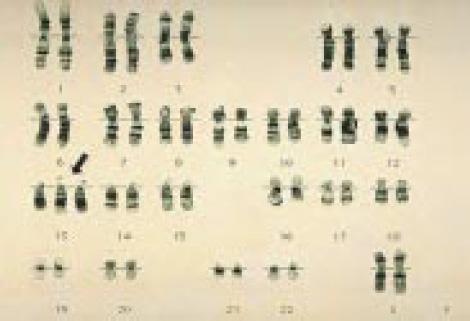
Хромосомы в случае синдрома Патау — Трисомия 13

Хромосомы в случае синдрома Эдвардса — Трисомия 18
Ранние признаки
В отличие от большинства заболеваний, связанных с нарушением количества хромосом, внутриутробное развитие детей с синдромом Клайнфельтера проходит нормально, склонности к преждевременному прерыванию беременности не наблюдается. Так что в младенческом и раннем детском возрасте заподозрить патологию практически невозможно. Более того, клинические признаки классического синдрома Клайнфельтера проявляются, как правило, только в подростковом периоде. Однако есть симптомы, которые позволяют заподозрить наличие синдрома Клайнфельтера в препубертатном периоде:
- высокий рост (пик прибавки роста приходится на период между 5–8 годами);
- длинные ноги (непропорциональное телосложение);
- высокая талия.
У части пациентов наблюдается некоторая задержка в развитии речи.
В подростковом возрасте синдром часто проявляется гинекомастией, которая при данной патологии имеет вид двустороннего симметричного безболезненного увеличения грудных желез. Так как такого рода гинекомастия часто наблюдается у совершенно здоровых подростков, этот симптом часто остается без внимания. В норме подростковая гинекомастия бесследно исчезает в течение нескольких лет, у пациентов же с синдромом Клайнфельтера обратной инволюции грудных желез не происходит. В некоторых случаях гинекомастия может не развиваться вовсе, и тогда патология проявляется признаками андрогенной недостаточности уже в постпубертатный период.
Причины отклонений, как они проявляются и какова вероятность возникновения
Рассмотрим несколько наиболее распространенных отклонений:
Синдром Дауна. В среднем встречается один родившийся с синдромом ребенок на 1000 здоровых. Причиной его является трисомия по 21 хромосоме. В норме этих хромосом должно быть две, а при данном отклонении их три. В жизни это заболевание проявляется в пороке сердца, лицевых изменениях, деформации грудной клетки, гиперподвижности суставов и ряде других признаков. Что касается умственной отсталости, то дебильность развивается лишь в 5%, имбецильность– в 75%, идиотия, или тяжелая степень умственной отсталости – в 20%.
- Хромосомный мозаицизм. Отклонение, при котором в организме растения, человека или животного есть генетически различающиеся клетки. Различия могут быть по причине анеуплоидии, которая была описана выше. Причинами могут быть мутации на ранних стадиях зародыша, неправильном расхождении хромосом при делении клеток, генотерапия. У человека данное нарушение может привести к следующим синдромам: Дауна, Кляйнфельтера, Шерешевкого-Тернера, Эдвардса. У животных и человека – к гермафродитизму.
- Транслокация. Вид мутации, при которой переходит перенос участка с одной на другую, не являющуюся ее парой, то есть негомологичную. Причиной является нарушение в молекуле ДНК. Частота среди людей по данному отклонению: 1 на 1300 человек.
- Синдром Эдвардса. Причиной является трисомия по 18 хромосоме. Частота встречаемости: 1 на 5000. Проявляется в низком весе новорожденного, аномалиях черепа, пороке сердца, умственной отсталости и нарушении некоторых структур мозга, сниженном мышечном тонусе.
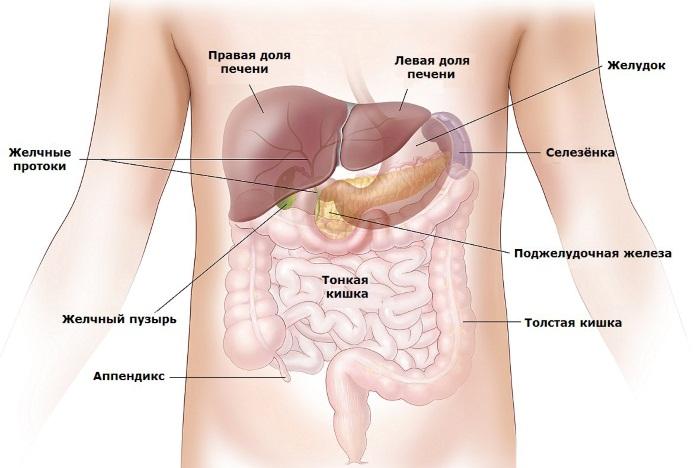
- Синдром Патау. Также является трисомией. Частота встречаемости 1:14000. При этом синдроме наблюдаются тяжелые врожденные пороки лицевой части черепа, лица, нарушение развития ЦНС, пороки сердца, нарушения поджелудочной железы, селезенки, высокая младенческая смертность.
- Синдром Шерешевского-Тернера. Основная причина – полное отсутствие второй пары. Затрагивает это нарушение только девочек. Встречаемость: 1 из 3000 новорожденных. Проявляется в недоношенности новорожденных, задержками речевого развития, пороками сердца, в короткой шее со складками кожи по бокам, отставании в физическом развитии.
Как воспитывать ребёнка с хромосомным заболеванием?
Воспитывать ребёнка с врождёнными хромосомными заболеваниями оказывается непросто. Для того чтобы облегчить свою жизнь, необходимо придерживаться некоторых правил. Во-первых, сразу следует преодолеть отчаяние и страх. Во-вторых, не нужно тратить время на поиске виновного, его просто нет
В-третьих, важно определиться с тем, какая помощь требуется ребёнку и семье, после чего обращаться к специалистам за медицинской и психолого-педагогической помощью
В первый год жизни диагностика крайне важна, так как в этот период развивается двигательная функция. С помощью профессионалов ребёнок быстрее приобретёт моторные способности. Необходимо объективно обследовать малыша на патологию зрения и слуха. Также ребёнок должен наблюдаться у педиатра, психоневролога и эндокринолога.
Родителям рекомендуется вступить в специальную Ассоциацию для того, чтобы получить ценные практические советы от людей, которые преодолели подобную ситуацию и готовы поделиться.

Носитель лишней хромосомы обычно дружелюбен, что облегчает его воспитание, также он по мере своих сил старается заслужить одобрение взрослого. Уровень развития особенного малыша будет зависеть от того, насколько упорно будут его обучать основным навыкам. Больные дети хоть и отстают от остальных, но требуют к себе много внимания. Всегда необходимо поощрять самостоятельность ребёнка. Прививать навыки самообслуживания следует на собственном примере, и тогда результат не заставит себя долго ждать.
Дети с хромосомными заболеваниями наделены особыми талантами, который необходимо раскрыть. Это могут быть занятия музыкой или рисование
Важно развиваться речь малыша, играть в активные и развивающие моторику игры, читать, а также приучать к режиму и аккуратности. Если проявить к ребёнку всю свою нежность, заботу, внимательность и ласку, он ответит тем же
Диагностика
Диагноз синдрома Тернера основан на идентификации характерных симптомов, детальной истории пациента, тщательной клинической оценке и различных специализированных тестов. Его следует подозревать у девочек с дефицитом роста или небольшим ростом неизвестной причины.
Клинические исследования
Диагноз подтверждается хромосомным анализом, который достигается путем определения кариотипа. Кариотипирование – лабораторный тест, оценивающий количество и структуру хромосом. Его можно проводить практически на любом типе ткани. В большинстве случаев используется образец крови для определения кариотипа человека.
Синдром Шерешевского Тернера определляется до рождения (пренатально) на основе генетического анализа, проведенного с помощью амниоцентеза или хорионной выборки ворсинок (CVS).
В некоторых случаях физические признаки заболевания можно увидеть на фетальном ультразвуке. Например, накопление лимфатической жидкости вблизи шеи развивающегося плода.
Конкретные методы визуализации, такие как магнитно-резонансная томография (МРТ), выполняется для обнаружения симптомов, потенциально связанных с синдромом Тернера, таких как нарушения функции печени, почек, сердца.
Многие люди с диагнозом проходят полную диагностику работы сердца, чтобы оценить его структуру и функцию. Диагностика включает эхокардиограмму.
Проводится дополнительная оценка функции щитовидной железы, печени, возраста костей, рост. Следует сделать скрининг на предмет наличия гипертензии.
Младенцы, диагностированные при рождении, должны пройти полное обследование уха, носа, горла, включая тесты на слух. Люди имеющие повторяющийся отит требуют периодической оценки слуха. Пострадавшие проходят тесты на функцию щитовидной железы из-за возможности ее заболевания.
Причины генетической патологии
ДНКнуклеотидовбелок, какой-либо фермент или рецептор организмаВсе хромосомы в организме человека делятся на два вида:
- Аутосомы. Аутосомы – это хромосомные пары с 1 по 22. Они несут большой объем генетической информации и могут быть различных размеров. При синдроме Дауна у больных наблюдается утроение аутосомы под номером 21.
- Половые хромосомы. Половые хромосомы обозначаются цифрами Х и Y. Они предопределяют пол человека (XX – девочка, XY – мальчик). Условно эти хромосомы объединяют в 23-ю пару, хотя Х и Y не похожи друг на друга ни размером, ни формой, ни набором генов.
в последовательности нуклеотидовдля женщиндля мужчиндве хромосомы, составляющие пару, соединяются не в виде буквы Х, а в виде буквы VВ зависимости от характера хромосомной мутации различают следующие виды болезни:
- Полная трисомия 21. Полная трисомия 21 предполагает, что у ребенка в каждой клетке организма имеется целая дополнительная хромосома. Таким образом, общее количество ее копий – 3. Частота данного варианта составляет 90 – 95%. Эта форма является наиболее тяжелой. У пациента наблюдается избыток всех генов, закодированных в этой молекуле ДНК. Как правило, нарушения внутриутробного развития у них встречаются чаще, а умственная отсталость более выражена. Полная трисомия возникает, если один из родителей передает ребенку не одну, а две хромосомы 21. Тогда при слиянии с третьей 21-й хромосомой (от второго родителя) возникает трисомия. Зигота (первая клетка, из которой возникает зародыш) уже содержит дефект. Дальнейшее ее деление объясняет, что все дочерние клетки будут похожи на нее.
- Мозаичная форма. При мозаичной форме механизм появления хромосомного дефекта несколько иной. Обе родительские гаметы (половые клетки) имели нормальное количество хромосом. После их слияния образовалась нормальная зигота с кариотипом 46, ХХ или 46, XY. В процессе деления этой первоначальной клетки ДНК распределилось неправильно. Часть клеток организма получилась с нормальным кариотипом, а часть – с кариотипом синдрома Дауна. Такая аномалия встречается довольно редко (3 – 5% случаев данного заболевания). Прогноз при ней лучше, так как здоровые клетки отчасти компенсируют генетический дефект. Ребенок все равно родится с синдромом Дауна и видимым отставанием в развитии. Однако выживаемость таких детей значительно выше. У них редко встречаются тяжелые пороки развития внутренних органов, несовместимые с жизнью.
- Семейный синдром Дауна. Семейный синдром Дауна – это весьма редкий генетический дефект (менее 2% случаев). При нем один из родителей имеет небольшие отклонения. Часть хромосомы 21 (а именно, критический участок) прикрепляется к другой хромосоме (обычно к 14-й). Таким образом, на 14 хромосоме содержится больше генетической информации, чем должно быть в норме. У человека при этом обычно нет видимых изменений (симптомов синдрома Дауна). Однако все половые гаметы, которые производит его организм, содержат этот дополнительный участок хромосомы 21. Очень высока вероятность, что в процессе образования зиготы такая гамета вызовет появление дополнительной 21-й хромосомы. Таким образом, дети у человека с подобным дефектом часто рождаются с синдромом Дауна. Из-за этой аномалии, передающейся потомству, данную форму болезни назвали семейной.
- Частичная трисомия 21. При частичной трисомии 21 у пациента обнаруживается не вся дополнительная хромосома, а лишь ее фрагмент с критическим участком. Из-за этого у ребенка развивается синдром Дауна в более легкой форме (однако все основные симптомы все равно присутствуют). Механизм такого дефекта чем-то похож на семейную форму болезни, но синдром не будет передаваться по наследству. Встречается этот вариант болезни очень редко.
На образование аномальных гамет могут повлиять следующие факторы:
- экологическая обстановка;
- некоторые медикаменты;
- курение;
- алкоголизм;
- радиация;
- некоторые заболевания половой сферы.
зачать ребенкаВероятность рождения ребенка в зависимости от возраста матери выглядит следующим образом:
- 0,064% для женщин, рожающих в возрасте 20 – 24 лет;
- 0,1% — для женщин в возрасте 25 – 30 лет;
- 0,17% — для женщин в возрасте 31 – 35 лет;
- 0,47% – для женщин 36 – 40 лет;
- 0,78% — для женщин 41 – 45 лет;
- до 5,25% — у женщин старше 45 лет (синдром Дауна – у каждого двадцатого ребенка).
Лечится ли синдром дауна
Лечение синдрома Дауна невозможно, но существуют программы поддержки и образования, которые могут помочь как людям с состоянием, так и их семьям.
Обычно программы начинаются с вмешательства в младенчестве. Учителя специального образования и терапевты помогают ребенку приобрести:
- сенсорные навыки;
- навыки общения;
- навыки самопомощи;
- двигательные навыки;
- язык и познавательные способности.
Дети с синдромом Дауна могут учиться медленнее, чем другие дети.
Школа является важной частью жизни ребенка, независимо от умственных способностей. Государственные и частные школы во многих странах поддерживают людей, их семьи специальными образовательными возможностями
Обучение дает ценную социализацию, помогает учащимся осваивать важные жизненные навыки
Государственные и частные школы во многих странах поддерживают людей, их семьи специальными образовательными возможностями. Обучение дает ценную социализацию, помогает учащимся осваивать важные жизненные навыки.
В течение последних 10 лет произошел резкий рост исследовательских усилий, направленных на терапевтические вмешательства для улучшения обучения и памяти.
Использовались три основные стратегии: имплантация нервных стволовых клеток; экологическое обогащение, физические упражнения, фармакотерапия.
Многие стратегии улучшают обучение и память, также электрофизиологические, молекулярные изменения у пораженных животных.
На сегодняшний день восемь молекул были протестированы в клинических испытаниях для взрослых людей.
До сих пор не проводились исследования для младенцев. Однако убедительные исследования показывают, что постоянные изменения головного мозга происходят во время роста плода.
Ранняя пренатальная диагностика дает окно на 28 недель, чтобы положительно повлиять на развитие мозга, улучшить постнатальный когнитивный эффект.
Несколько подходов (эпигаллокатехин галлат, NAP / SAL, флуоксетин, апигенин) были использованы для лечения мышей внутриутробно; они показали терапевтические эффекты, которые сохранялись до взрослой жизни.